Just Write Your Own Python Parsers for .fastq Files
Posted on Thu 22 August 2019 in commentary
In contrast to the zen of python there are actually many ways to handle sequence data in Python. There are several packages on PyPI that provide parsers for sequence formats like .fastq and .fasta. I've never bothered with these, including the oft-used Biopython. I vaguely remembered Biopython being slower than any parser I'd written myself early-on in learning bioinformatics, and it not actually being simpler to implement. However, I'd never looked at this in detail. Additionally, I'd recently run across a few posts on biostars where users were deriding people for asking "What is the most efficient way to parse a huge .fastq file" for something similar.
First of all, don't discourage people who are trying to learn. Secondly, this is a good question! As scientists, we should know that just because data exists doesn't meant it's good. Likewise, just because software exists doesn't mean it's the best tool for any given job. Plus, writing simple parsers for common formats is a good way to practice file processing for when you eventually need to do something hard and no ready-made parser exists in a package.
Rather than vaguely saying "X package is slow, do this instead" I thought it'd be best to actually benchmark some different .fastq parser options.
The Contenders
There are several packages that include parsers for biological sequence data. These include:
I'm familiar with Biopython from the recommendations that abound in the community
for exactly this task, and HTSeq mostly for HTSeq-count. Scikit-bio seems to be
newer and under current development, so results from testing that are subject to
change. Just in case someone looks at this yers after it's written and wonders
why I got the performance that I did.
When it comes to dealing with .fastq files I checked through my library of Python scripts and came across two patterns that I'll also test compared to these packages:
- Reading line-by-line, using a counter to yield records
- Reading line-by-line, using
zip_longest()fromitertoolsto yield records
Setting up the Test
I did this in a jupyter notebook, since that's what I use on a day-to-day basis. Most of my interactive "data science" work is done in R, which is mostly a consequence of at one point needing to use some R packages that have no Python equivalents, and just rolling with that. So, actually using Python in jupyter is a bit of a departure from the norm for me.
First, you need the necessary packages. I just use pip with pyenv:
pip install biopython HTSeq scikit-bio
Then, started a new jupyter notebook with jupyterlab (a sweet new UI for jupyter that you should use!). Your first step is always to do your imports.
from Bio import SeqIO
from HTSeq import FastqReader
from itertools import zip_longest
import skbio
I'm only using one function from skbio, but it's just called read() which is
too generic a name to just import that single function without causing all sorts
of annoyances and gnashing of teeth.
Also, it's important with any parsing problem to understand the file format. The .fastq format is ubiquitous in bioinformatics and looks like this:
@SEQ_ID
GATTTGGGGTTCAAAGCAGTATCGATCAAATAGTAAATCCATTTGTTCAACTCACAGTTT
+
!''*((((***+))%%%++)(%%%%).1***-+*''))**55CCF>>>>>>CCCCCCC65
You can understand it as a repeated series of four lines:
- Sequence ID, starting with "@"
- Sequence (ATCG)
- Separator (+)
- Quality score for each base call (same length as sequence)
The catch here is that you can't use @ as a record separator. It's a valid character in the score line, too. So, you really do need to group the lines in batches of four, as it's possible @ will exist in position 1 of the score line.
Define Some Functions to Test
In order to make the benchmarking easier to follow, I figured I'd define the functions I want to bechmark in a consistent way:
# Using Biopython
def parse_biopython(input_fastq):
for record in SeqIO.parse(input_fastq, 'fastq'):
yield record
# Using HTSeq
def parse_htseq(input_fastq):
for record in FastqReader(input_fastq):
yield record
# HTSeq raw
def parse_htseq_raw(input_fastq):
for record in FastqReader(input_fastq, raw_iterator=True):
yield record
# Skbio
def parse_skbio(input_fastq):
for record in skbio.io.read(input_fastq, format='fastq'):
yield record
# Line by line with counter
def parse_lbl_counter(input_fastq):
with open(input_fastq, 'r') as input_handle:
record = []
n = 0
for line in input_handle:
n += 1
record.append(line.rstrip())
if n == 4:
yield record
n = 0
fq_record = []
# Line by line with zip_longest
def parse_zip_longest(input_fastq):
with open(input_fastq, 'r') as input_handle:
fastq_iterator = (l.rstrip() for l in input_handle)
for record in zip_longest(*[fastq_iterator] * 4):
yield record
Here I intended to use two different methods from HTSeq, one which just returns
bare tuples rather than objects with other kinds of validation based on the
definition of the format. However, neither HTSeq method worked. Instead, giving a
StopIteration error when it reached the end of a file. Trying to catch that
with a try: except: block didn't seem to work? It did parse until it reached
the end of a file though. I think this is a bug, and I may raise it with the
HTSeq people. So it is, regrettably, not included in my benchmarking results.
Also, in both custom parsers, str.rstrip() was marginally faster than
str.strip() so I went with that instead.
Run Some Benchmarks
I decided I would try each of these with 1 million lines from a whole-genome bisulfite experiment. These are the R1 mates from 75bp paired end reads:
%timeit -n 10 -r 10 [record for record in parse_biopython('JWG3_2_2_R1.head.fastq')]
2.86 s ± 56.7 ms per loop (mean ± std. dev. of 10 runs, 10 loops each)
%timeit -n 10 -r 10 [record for record in parse_skbio('JWG3_2_2_R1.head.fastq')]
1min 33s ± 13.7 s per loop (mean ± std. dev. of 10 runs, 10 loops each)
%timeit -n 10 -r 10 [record for record in parse_lbl_counter('JWG3_2_2_R1.head.fastq')]
295 ms ± 14.7 ms per loop (mean ± std. dev. of 10 runs, 10 loops each)
%timeit -n 10 -r 10 [record for record in parse_zip_longest('JWG3_2_2_R1.head.fastq')]
249 ms ± 2.57 ms per loop (mean ± std. dev. of 10 runs, 10 loops each)
The %timeit function there is some ipython "line magic." It simplifies timing
a single line of code. The %%timeit is the "cell magic" version.
It seems that skbio isn't ready for primetime just yet. The real question then
is, would biopython suffice for day-to-day work? Perhaps yes, ~1M lines in < 3s
(349650.35 lines per second) is a timescale that people might be willing to work
with. Keep in mind this is on my personal laptop, so it's hardly a compute
cluster. In contrast, the very simple line counter-based parser that I wrote as a
master's student back in 2013 as a python-learning exercise is nearly 10x faster!
There is also an improvement in speed for using zip_longest() from itertools
(a trick I'm pretty sure I saw in a post from Brent Pedersen on stackoverflow).
Visualize
I'm usually a ggplot2 useR for visualizations, but I'm already in python here so let's use this as an excuse to try the great python plotting library altair. It's declarative, like ggplot2, and you build your plot by "mapping" your "variables" (columns) to "encodings" (analogous to "aesthetics" in ggplot2). I ran several other benchmarks and turned them into a pandas data frame. First you'll need to do some imports:
import pandas as pd
import numpy as np
import altair as alt
Then make the data frame
# Create a dataframe Pandas style
timing_data = pd.DataFrame({'Method': np.repeat(['biopython', 'skbio', 'lbl', 'zip'], 5),
'Reads': (np.tile([100, 1000, 10000, 100000, 1000000], 4) / 4),
'Time (s)': [(670 / 1e6), (4.4 / 1000), (40.49 / 1000), (418 / 1000), 2.86,
(14.2 /1000), (132 / 1000), 1.32, 13.9, (60 + 33),
(181 / 1e6), (442 / 1e6), (3.92 / 1000), (40.5 / 1000), (295 / 1000),
(70.2 / 1e6), (352 / 1e6), (3.19 / 1000), (32.5 / 1000), (249 / 1000)]})
Since each record is 4 lines, converting lines to # of reads requires dividing by four. Likewise, the benchmarking results are in various time units, so I've converted all of them to seconds. Not particular efficiently, but for this simple example it's fine.
Now we can visualize with Altair. It has a very nice syntax inspired by ggplot2's "grammar of graphics." It's based on vega-lite under the hood and allows you to easily save your plot from jupyterlab. Here's the code:
# Plot as a scatterplot
alt.Chart(timing_data).mark_point().encode(
x='Reads',
y='Time (s)',
color='Method'
)
# Plot on log scale
alt.Chart(timing_data).mark_point().encode(
alt.X('Reads', scale=alt.Scale(type='log', base=10)),
alt.Y('Time (s)', scale=alt.Scale(type='log', base=10)),
color='Method'
)
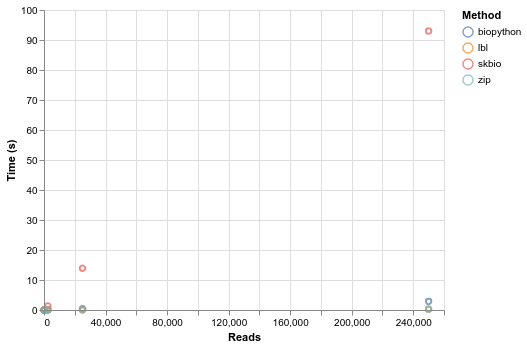

Everything scales linearly, but at massively different rates. Sci-kit bio is in another universe in terms of time, such that you can't even visualize it with the others in a meaningful way until you log scale everything. By the log scale, you can essentially see that biopython is an order of magnitude faster than skbio, and either simple parser are an order of magnitude faster again. The difference between the two simple parsers is pretty insignificant.
Note: Altair is great! Not quite as full-featured as ggplot2 in R, but it's definitely promising and something to watch for in the future. They definitely should make it work with jupyterlab's dark theme though. Due to the transparent plot backgrounds it requres a light theme.
To Wrap Things Up
I'm not saying you should never use biopython, I suspect its parser does some
extra validation that my simple parsers don't. It also returns objects with some
possibly useful methods. However, if you just want to read files quckly then the
simple line-by-line parsers aren't actually very complicated to write. Plus, you
don't even need to import anything unless you want a minor speed boost from
itertools. Additionally, if you didn't need to strip newlines you'd get a boost
from not calling an str.strip() method on each line.
If you're ok with living dangerously, and are sure your files are formatted correctly you can easily write something that will outperform standard implementations with little effort when it comes to .fastq parsing.